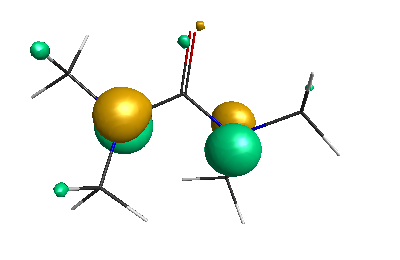http://www8.ocn.ne.jp/~ymakioka/
[GAMESS] GAMESSでかんたん(?)分子軌道計算…その1:インストールから計算実行まで。ただしMac版。
http://kyoroski.blogspot.com/2010/08/gamess-gamess1mac.html
DFT-geometrical optimization of N,N-dimethylformamide (N,N-ジメチルホルムアミド) was performed at the B3LYP/6-31G(d) level. Then energy levels were calculated at the same level.
Content of the input file from here -----
! DFT, B3LYP/6-31G(d), optimization
$CONTRL DFTTYP=B3LYP SCFTYP=RHF RUNTYP=OPTIMIZE $END
$SYSTEM TIMLIM=600000 MEMORY=80000000 $END
$STATPT OPTTOL=0.0001 NSTEP=200 PROJCT=.FALSE. $END
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$SCF DIRSCF =.TRUE. $END
$DATA
C1
C 6.0 -5.0936614568 2.5180700697 0.0011616408
O 8.0 -6.2791308734 2.2134708972 0.0004586203
N 7.0 -4.0309774156 1.6395202249 -0.0059655718
H 1.0 -4.7765742690 3.5738923224 0.0071047561
C 6.0 -2.6600686301 2.1086173994 0.0045544996
C 6.0 -4.2578351320 0.2077910434 -0.0114215304
H 1.0 -2.6165063429 3.2014277751 0.0024082472
H 1.0 -2.1487018777 1.7278689332 -0.8837721631
H 1.0 -2.1644733589 1.7322990160 0.9036786396
H 1.0 -5.3249632762 -0.0324263825 -0.0224603790
H 1.0 -3.8052330359 -0.2242840528 0.8854295083
H 1.0 -3.7875892300 -0.2205069957 -0.9009552297
$END
to here -----
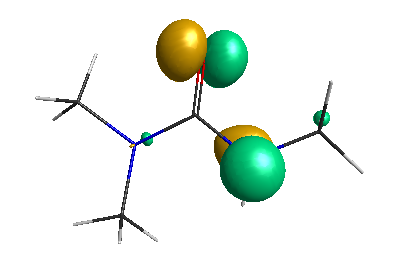
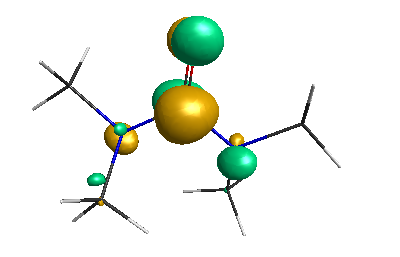
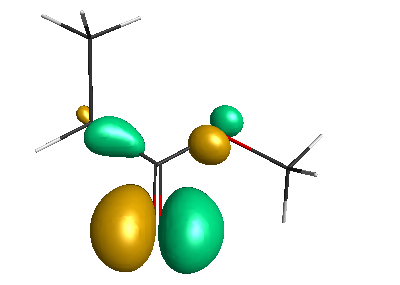
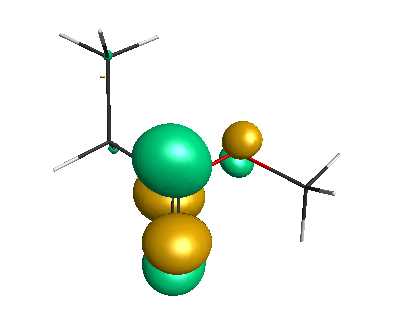
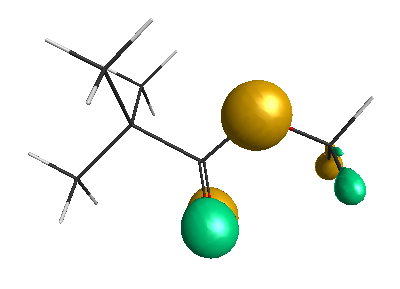
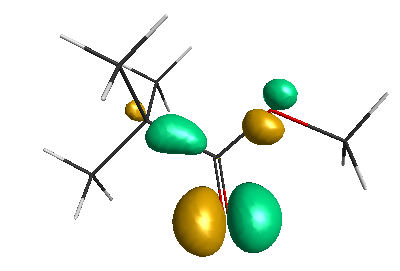
Results of geometrical optimization...
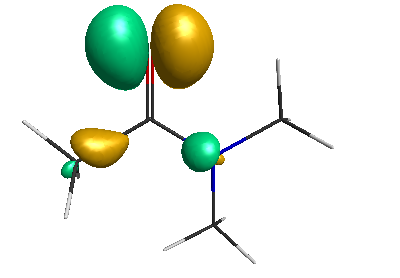
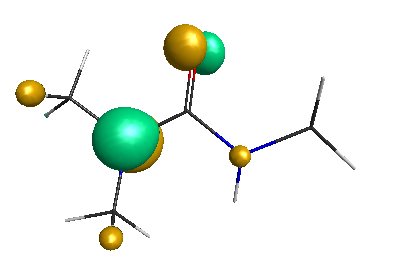
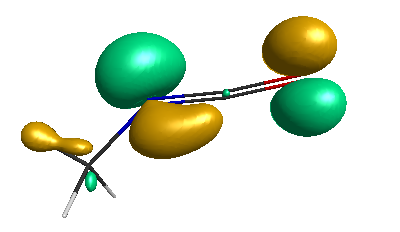
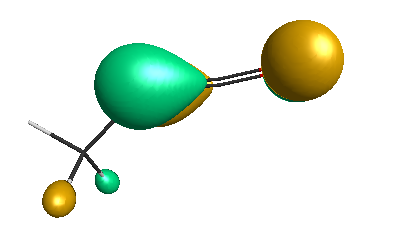
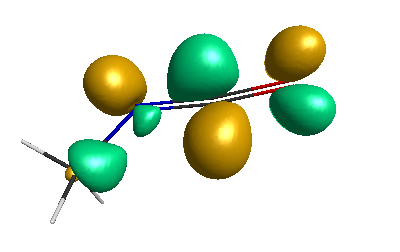
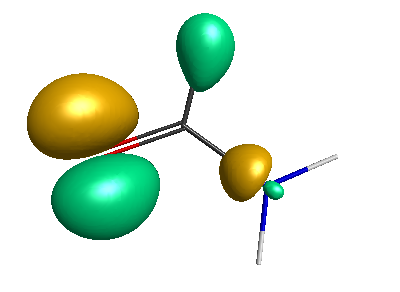
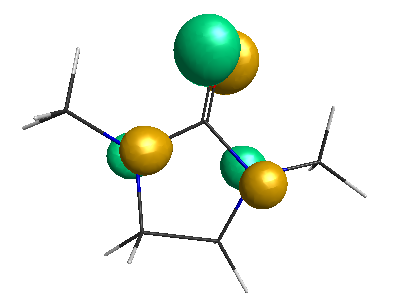
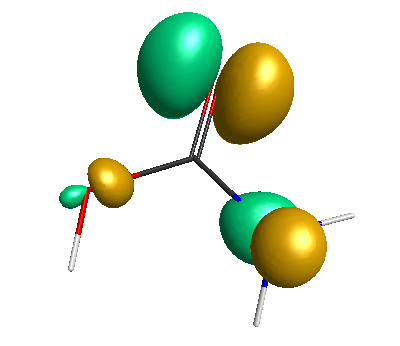
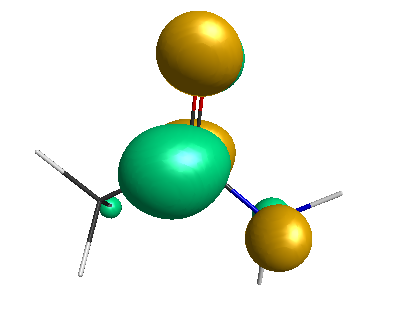
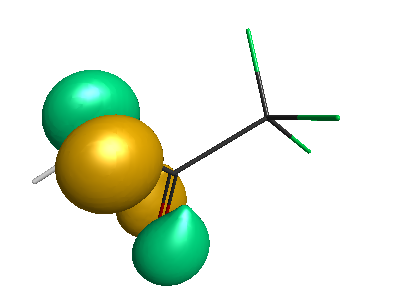
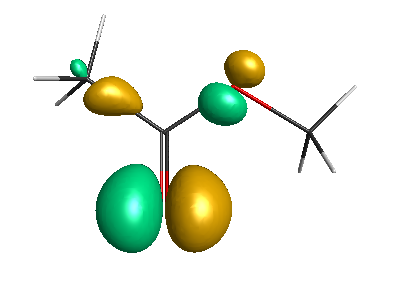
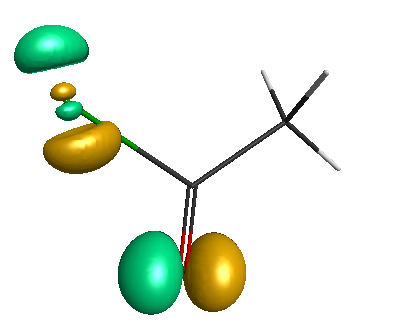
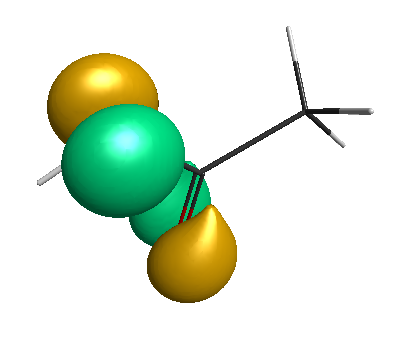
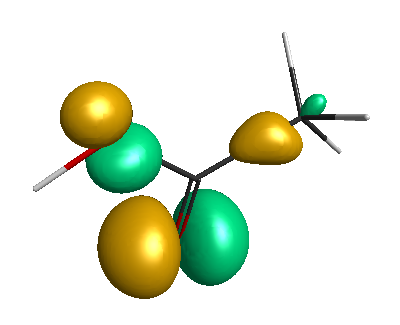
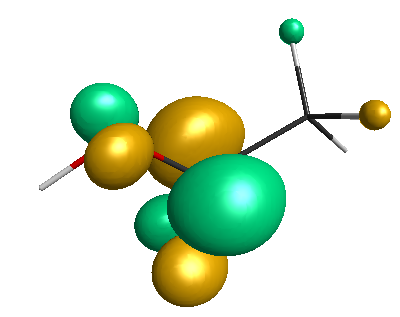
HOMO-1 (-6.75 eV):
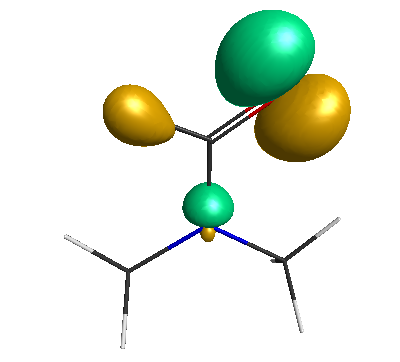
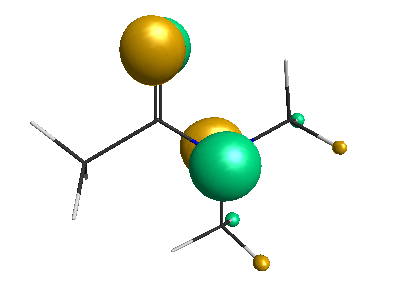
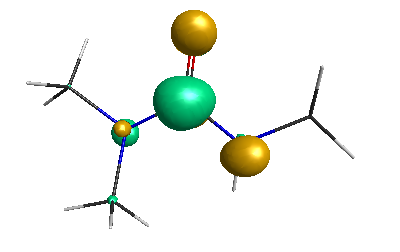
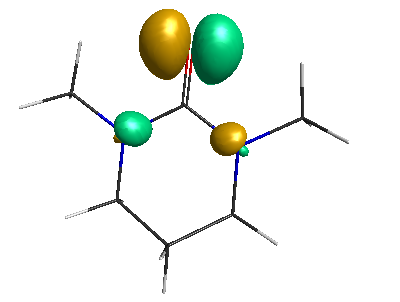
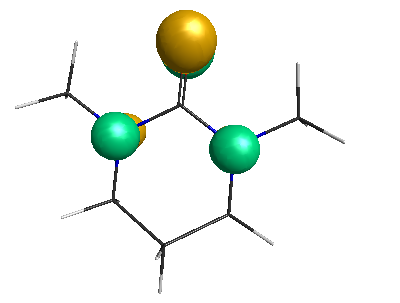
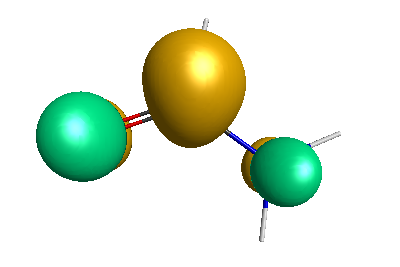
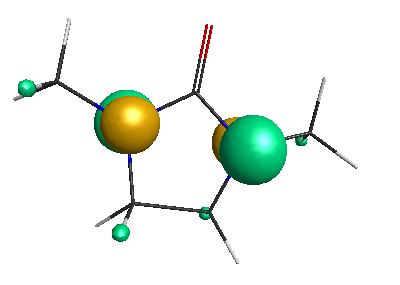
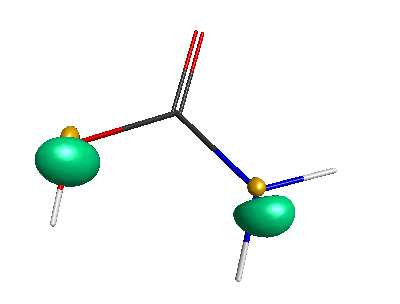
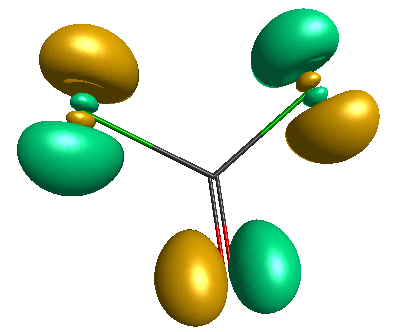
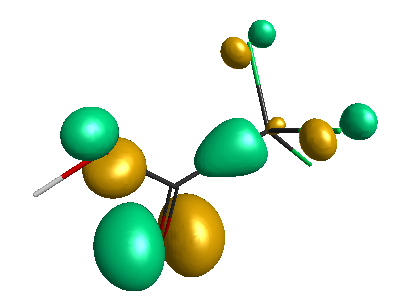
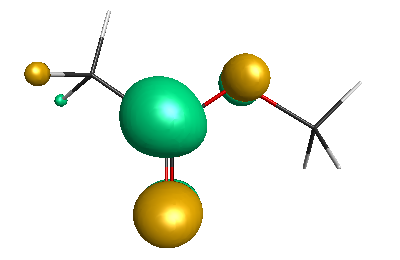
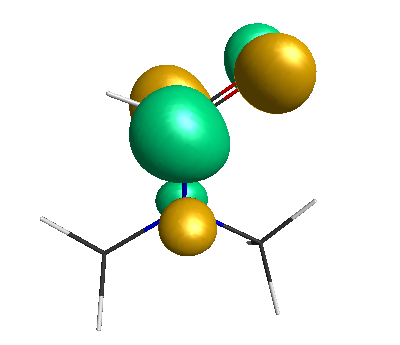
HOMO (-6.50 eV):
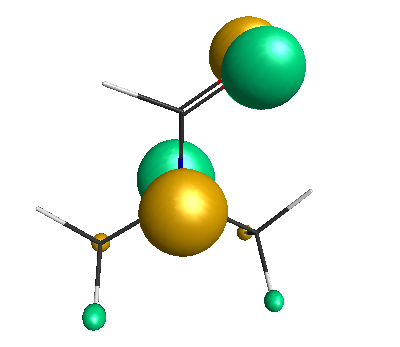
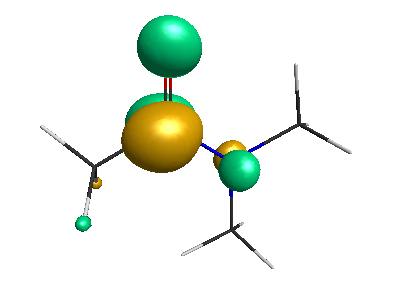
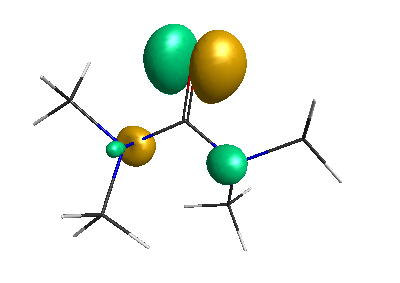
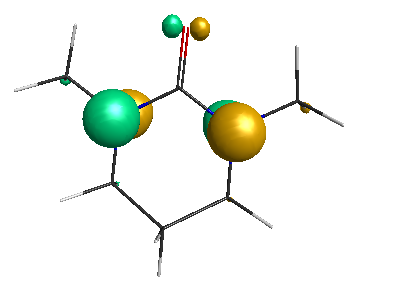
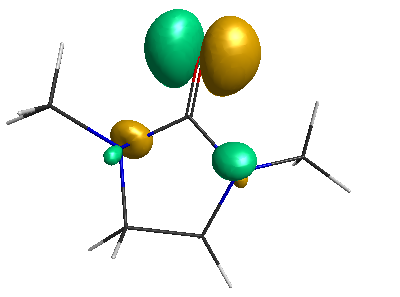
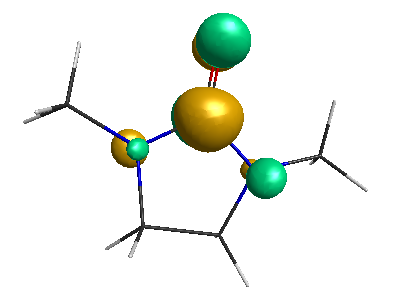
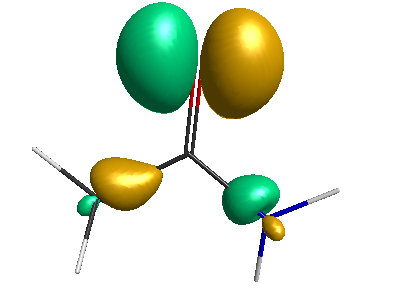
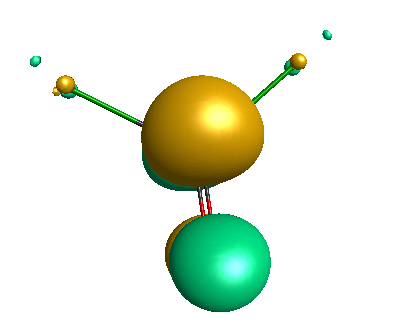
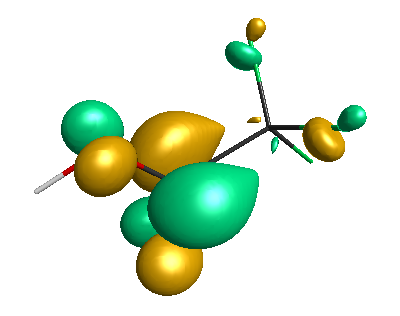
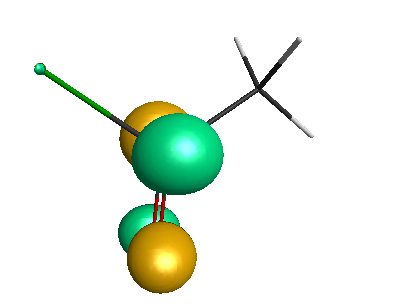
LUMO (+0.98 eV):
Molecule modeling: Avogadro
http://avogadro.openmolecules.net/wiki/Main_Page
Geometry optimization and energy calculation: GAMESS-US
http://www.msg.chem.iastate.edu/gamess/
Structure display: MacMolPlt
http://www.scl.ameslab.gov/MacMolPlt/