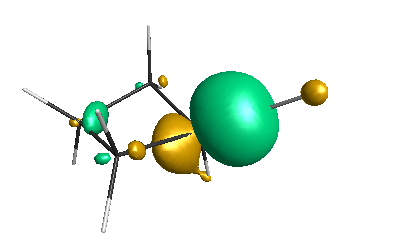http://www8.ocn.ne.jp/~ymakioka/
[GAMESS] GAMESSでかんたん(?)分子軌道計算…その1:インストールから計算実行まで。ただしMac版。
http://kyoroski.blogspot.com/2010/08/gamess-gamess1mac.html
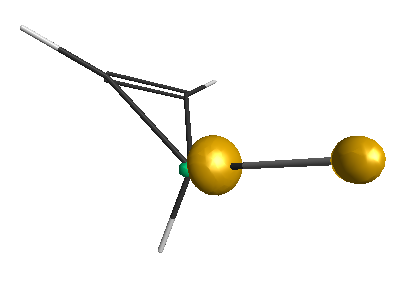
DFT-geometrical optimization of xx was performed at the B3LYP/6-31G(d) level. Then energy levels were calculated at the same level.
Content of the input file from here -----
! DFT, B3LYP/6-31G(d), optimization
$CONTRL DFTTYP=B3LYP SCFTYP=RHF RUNTYP=OPTIMIZE $END
$SYSTEM TIMLIM=600000 MEMORY=80000000 $END
$STATPT OPTTOL=0.0001 NSTEP=100 PROJCT=.FALSE. $END
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$SCF DIRSCF =.TRUE. $END
$DATA
C1
C 6.0 -1.3360887702 0.2666015109 -0.8040335260
C 6.0 0.0277917463 0.8225020068 -1.1008366430
C 6.0 -1.1877534731 1.6918584653 -1.2556161328
Li 3.0 -1.8129703686 -0.0635706993 1.0891041480
H 1.0 -1.7577236859 -0.4469914652 -1.5021946111
H 1.0 -1.3483583153 2.4763294201 -0.5250981989
H 1.0 -1.5089401493 1.9434324079 -2.2596093740
H 1.0 0.5297698571 0.4852101101 -2.0004624404
H 1.0 0.6903013617 1.0180891890 -0.2659409358
$END
to here -----
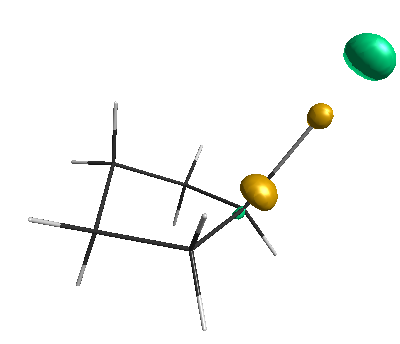
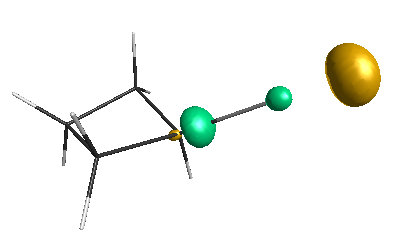
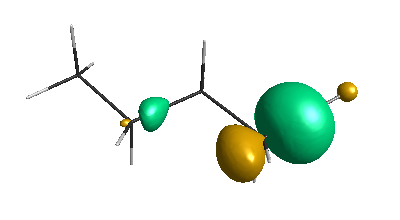
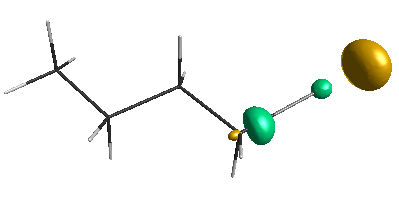
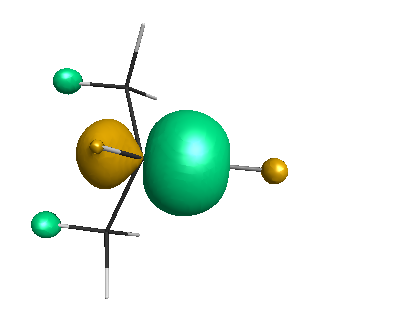
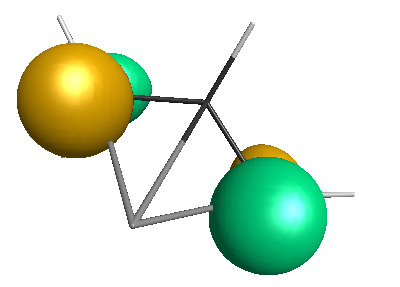
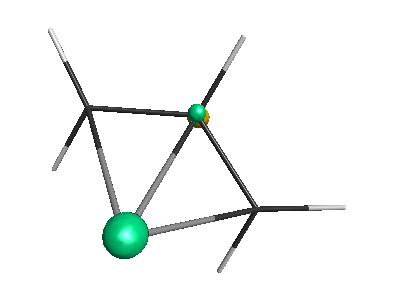
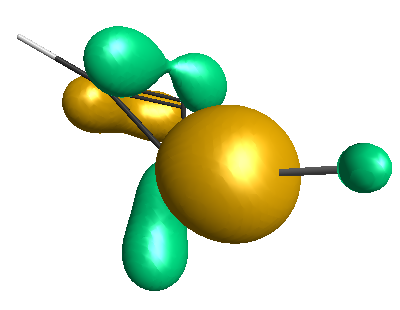
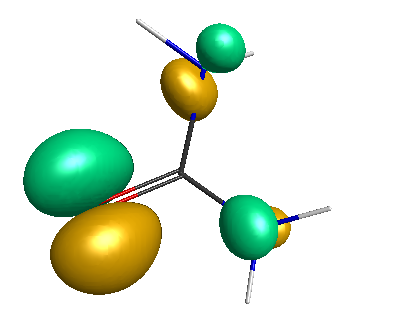
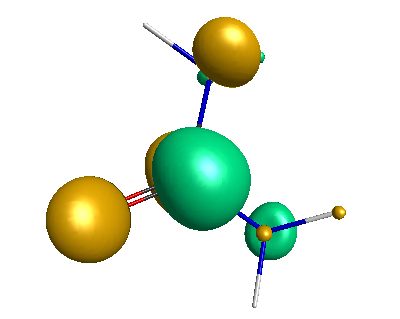
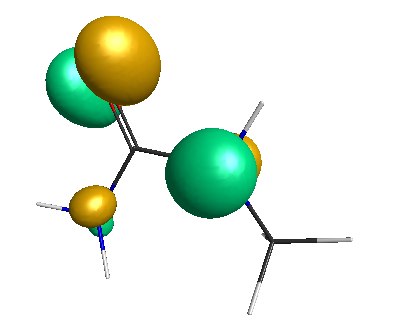
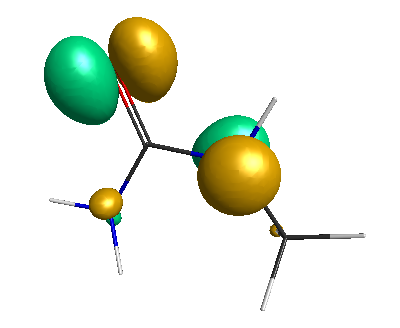
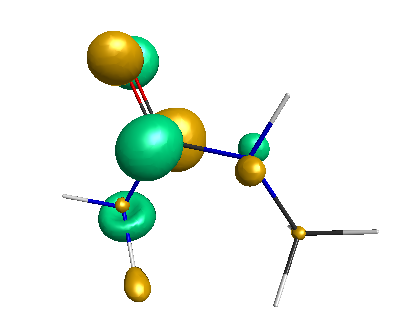
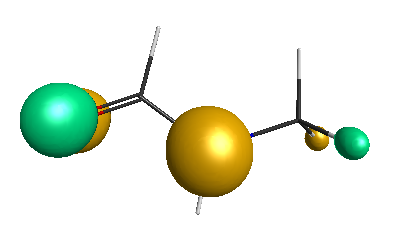
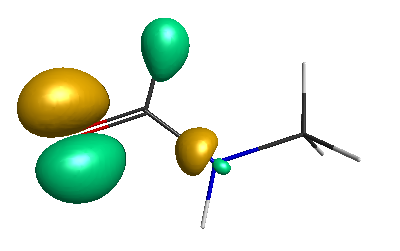
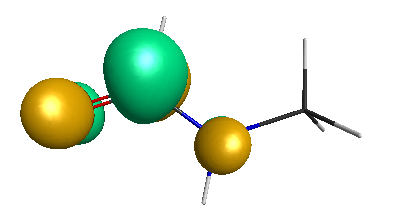
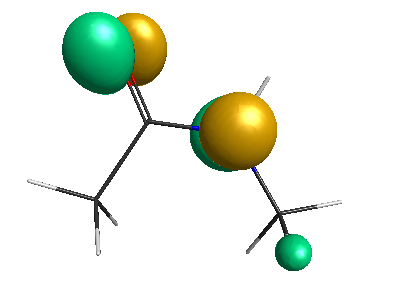
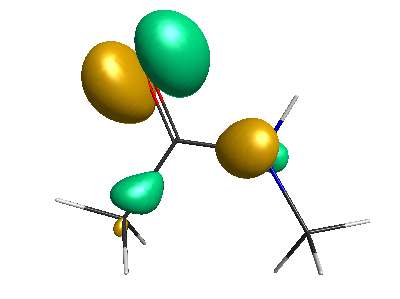
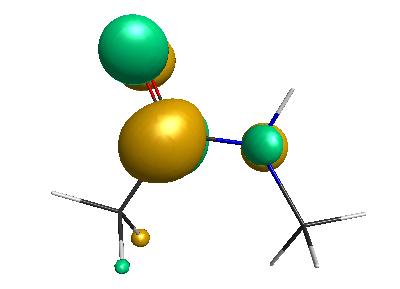
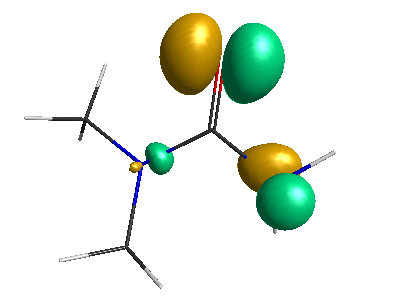
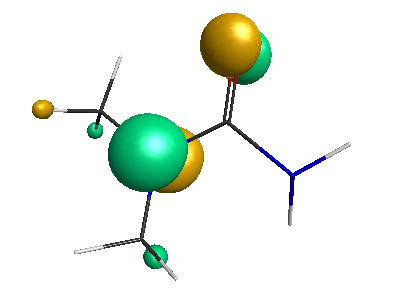
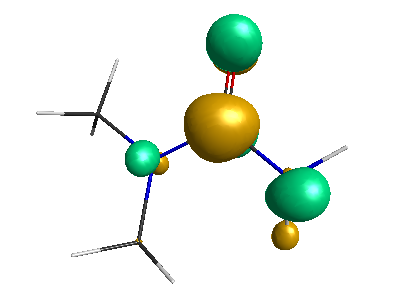
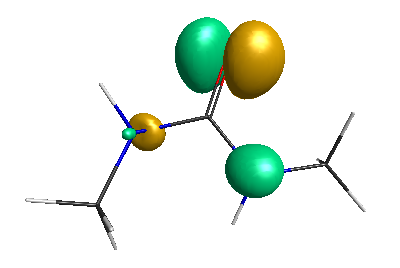
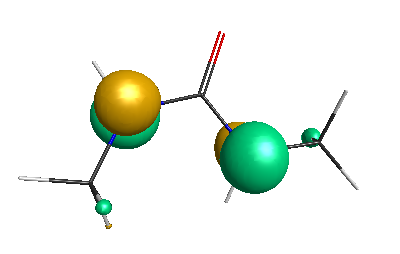
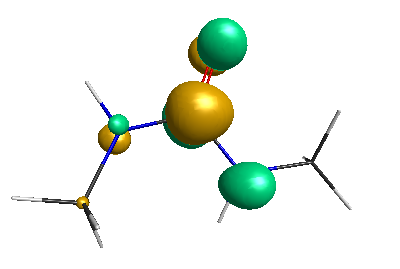
Results of geometrical optimization...
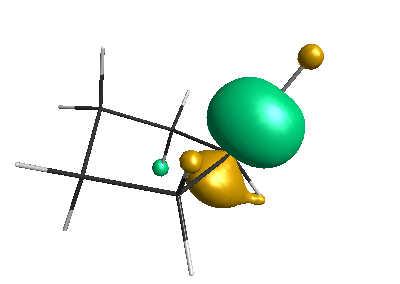
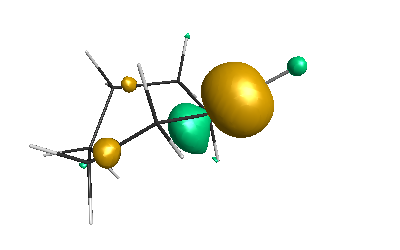
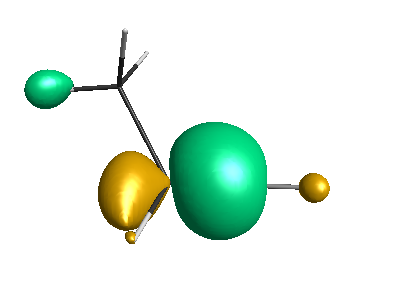
HOMO (-4.46 eV):
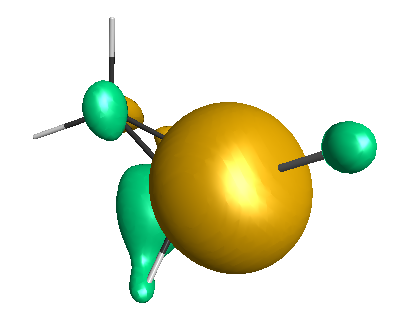
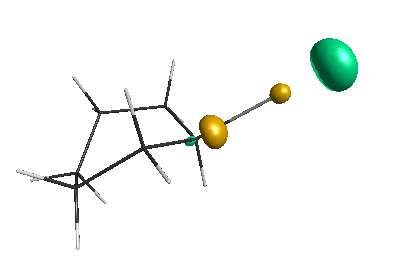
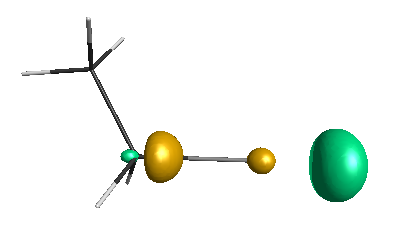
LUMO (-1.20 eV):
Molecule modeling: Avogadro
http://avogadro.openmolecules.net/wiki/Main_Page
Geometry optimization and energy calculation: GAMESS-US
http://www.msg.chem.iastate.edu/gamess/
Structure display: MacMolPlt
http://www.scl.ameslab.gov/MacMolPlt/