http://www8.ocn.ne.jp/~ymakioka/
[GAMESS] GAMESSでかんたん(?)分子軌道計算…その1:インストールから計算実行まで。ただしMac版。
http://kyoroski.blogspot.com/2010/08/gamess-gamess1mac.html
DFT-geometrical optimization of 1,3-dilithiopropyne (1,3-ジリチオプロピン) was performed at the B3LYP/6-31G(d) level. Then energy levels were calculated at the same level.
Content of the input file from here -----
! DFT, B3LYP/6-31G(d), optimization
$CONTRL DFTTYP=B3LYP SCFTYP=RHF RUNTYP=OPTIMIZE $END
$SYSTEM TIMLIM=600000 MEMORY=80000000 $END
$STATPT OPTTOL=0.0001 NSTEP=100 PROJCT=.FALSE. $END
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$SCF DIRSCF =.TRUE. $END
$DATA
C1
C 6.0 -4.9604145102 2.6112470820 -0.0132957273
C 6.0 -3.7651447390 2.7252983110 -0.0098426375
C 6.0 -6.4171165747 2.4722712300 -0.0184543286
Li 3.0 -1.8538341282 2.9076687124 -0.0041167658
Li 3.0 -7.2671735540 4.1937176550 0.0021693910
H 1.0 -6.7575062888 1.9154553650 0.8610151238
H 1.0 -6.7540648557 1.9371796824 -0.9101504886
$END
to here -----
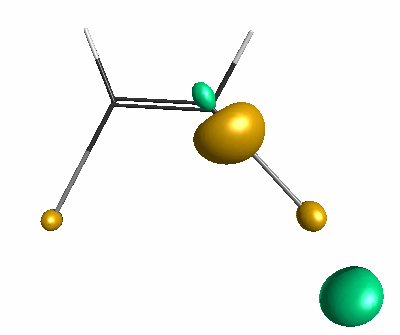
Results of geometrical optimization...
HOMO (-3.46 eV):

LUMO (-1.13 eV):

Molecule modeling: Avogadro
http://avogadro.openmolecules.net/wiki/Main_Page
Geometry optimization and energy calculation: GAMESS-US
http://www.msg.chem.iastate.edu/gamess/
Structure display: MacMolPlt
http://www.scl.ameslab.gov/MacMolPlt/