今回は1,2-ジシラベンゼンの構造最適化。いつものようにDFT, B3LYP/6-31G(d)。
-----ここから入力ファイルの中身
! DFT, B3LYP/6-31G(d), optimization
$CONTRL DFTTYP=B3LYP SCFTYP=RHF RUNTYP=OPTIMIZE $END
$SYSTEM TIMLIM=600000 MEMORY=80000000 $END
$STATPT OPTTOL=0.0001 NSTEP=200 PROJCT=.FALSE. $END
$BASIS GBASIS=N31 NGAUSS=6 NDFUNC=1 $END
$GUESS GUESS=HUCKEL $END
$SCF DIRSCF =.TRUE. $END
$DATA
C1
Si 14.0 -3.3763774280 4.1953130587 -0.2691731924
C 6.0 -4.6432146445 2.8882905836 -0.0542660182
C 6.0 -4.3805567575 1.5025156022 0.0012395544
C 6.0 -3.1026261338 0.8570143128 -0.0010040688
C 6.0 -1.8311215742 1.4637626318 0.0517465404
Si 14.0 -1.5228392153 3.2595591234 0.2604887214
H 1.0 -5.6707041799 3.2375791444 -0.0236717931
H 1.0 -5.2518680888 0.8406707450 0.0369803651
H 1.0 -3.1212841662 -0.2387008280 -0.0349797939
H 1.0 -0.9410990836 0.8417517220 0.0211479979
H 1.0 -3.6493850132 5.4899809715 0.4094561906
H 1.0 -0.3209969347 3.8018824940 -0.4267960642
$END
ここまで-----
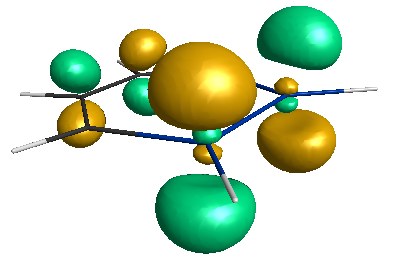
↓HOMO (-5.41 eV)。分子の平面性が保たれています。さすがに正六角形ではないですが…。モノシラベンゼン(-5.63 eV)より準位が高いですね。

↓LUMO (-1.03 eV)。モノシラベンゼン(-0.63 eV)よりも準位が低く、結果、HOMO-LUMOギャップが小さくなっています。
0 件のコメント:
コメントを投稿